| Table of Contents |  |
|
Case Report
| ||||||
| Intrahepatic cholangiocarcinoma with congenital hepatic fibrosis | ||||||
| Adeel S. Zubair1, Ali Metwaly2, Justin M. Burns3, John A. Stauffer4 | ||||||
|
1Medical Student, Mayo Medical School, Rochester, Minnesota, USA.
2Observing Physician, Mayo Clinic Jacksonville, Jacksonville, Florida, USA. 3Surgeon, Department of Transplantation, Mayo Clinic Jacksonville, Jacksonville, Florida, USA. 4Surgeon, Department of Surgery, Mayo Clinic Jacksonville, Jacksonville, Florida, USA. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] |


| How to cite this article: |
| Zubair AS, Metwaly A, Burns JM, Stauffer JA. Intrahepatic cholangiocarcinoma with congenital hepatic fibrosis. Int J Hepatobiliary Pancreat Dis 2015;5:103–107. |
|
Abstract
|
|
Introduction:
Little is known regarding the association between congenital hepatic fibrosis (CHF) and cholangiocarcinoma and publications reporting these associations are rare. In literature, only four cases reporting the associations exist; those four cases were all diagnosed at autopsy. Herein, we describe a case of CHF and intrahepatic cholangiocarcinoma that was successfully treated with surgical resection.
Case Report: A 46-year-old male with past medical history significant for CHF for 12 years, complicated by portal hypertension and esophageal varices, was found to have lesions on MRI suspicious for intrahepatic cholangiocarcinoma. The patient had relatively preserved liver function, and the decision was made to proceed with surgical resection. Pathology showed poorly differentiated cholangiocarcinoma with background features of CHF. Conclusion: All patients with CHF patients should be monitored for the development of cholangiocarcinoma. If the patient has compensated disease, treatment via partial hepatectomy is preferred. With vigilant screening and appropriate treatment, it is possible to decrease morbidity and mortality from the development of intrahepatic cholangiocarcinoma in the setting of CHF. | |
|
Keywords:
Cholangiocarcinoma, Cirrhosis, Congenital hepatic fibrosis
| |
|
Introduction
| ||||||
|
Congenital hepatic fibrosis (CHF) is an autosomal recessive disease that primarily affects the hepatobiliary and renal systems [1]. It is characterized by hepatic fibrosis, portal hypertension, and renal cystic disease. Pathologically, it is defined by its variable degree of periportal fibrosis and irregularly shaped proliferating bile ducts [2] [3]. It is one of the fibropolycystic diseases, which also include Caroli disease, autosomal dominant polycystic kidney disease, and autosomal recessive polycystic kidney disease. It results from a malformation of the ductal plate (the embryological precursor of the biliary system), secondary biliary strictures, and periportal fibrosis [4] subsequently, leading to the development of portal hypertension. It is a rare disease, and its exact incidence and prevalence are not known. Only a few hundred patients with CHF have been reported in literature [5]. Cholangiocarcinoma is an adenocarcinoma of the bile ducts that forms in the biliary epithelial cells and can be sub-classified into intrahepatic and extrahepatic bile duct cancer [6]. Intrahepatic cholangiocarcinoma (ICC) represents a tumor mass within a hepatic lobe or at the periphery of the liver [7]. Risk factors for cholangiocarcinomas are related to chronic inflammation of the biliary tract such as primary sclerosing cholangitis in Western countries and liver fluke infestation and hepatolithiasis (recurrent pyogenic cholangitis) in Asian countries. Other risk factors include Thorotrast exposure and biliary malformations such as choledochal cysts, Caroli disease, CHF, polycystic disease, and von Meyenburg complexes [7] [8]. Little is known regarding the association between CHF and cholangiocarcinoma and publications reporting these associations are rare [9]. In literature, only four cases reporting the associations exist; those four cases were all diagnosed at autopsy. Herein, we describe a case of CHF and ICC that was successfully treated with surgical resection. | ||||||
|
Case Report
| ||||||
|
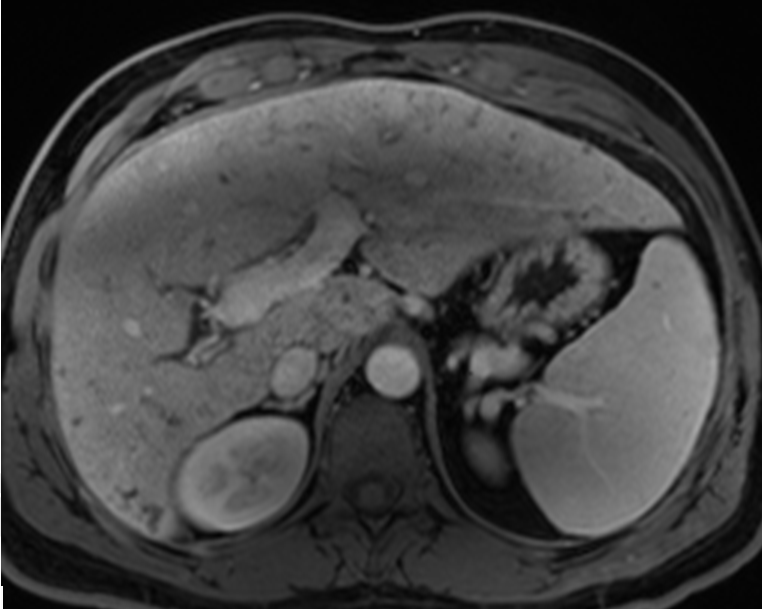
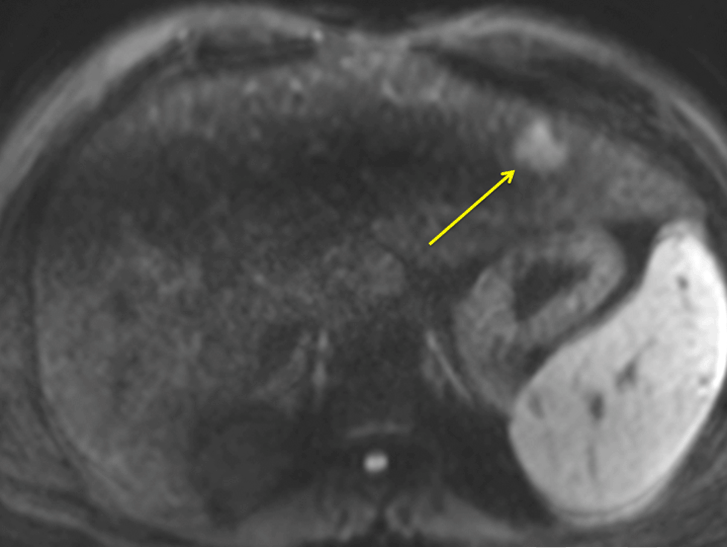
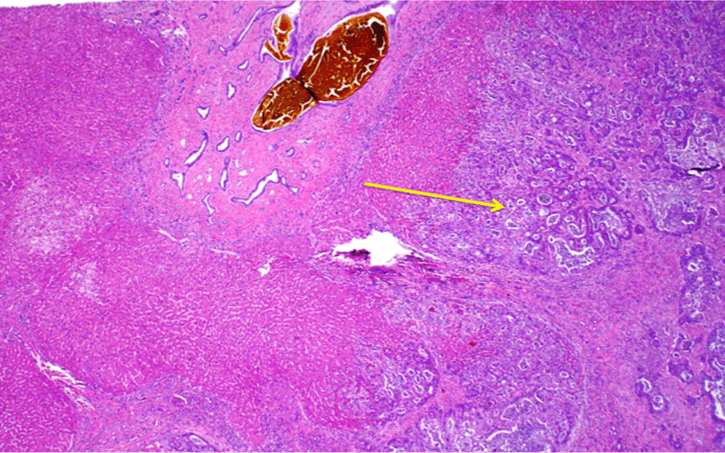
A 46-year-old male with past medical history significant for CHF complicated by portal hypertension and esophageal varices came to the hepatology clinic for routine follow-up. The patient had previously been diagnosed with CHF 12 years prior after an incidental finding of elevated liver function tests. Comprehensive workup including laboratory and serologic investigation, high quality cross sectional imaging, and liver biopsy ruled out viral, autoimmune, congenital, or any other etiology of liver disease. Liver biopsy at the time of diagnosis showed stage 4 fibrosis with numerous irregular bile ducts and narrow bile ductules lining the fibrous septa. The unremarkable parenchyma, absence of vasculature within the fibrous regions, and lack of other abnormal findings led to a diagnosis most compatible with CHF. During the visit, the patient reported weight loss with no other constitutional symptoms present. Blood work showed increased AST and ALT (45 u/L, 52 u/L) as well as CA 19–9 (317 u/mL). In light of the elevated CA-19-9, an MRI was performed to screen for the possibility of cholangiocarcinoma. The MRI scan showed cirrhotic hepatic morphology with severe fibrotic changes (Figure 1). No obvious lesions were seen within the liver except the diffusion weighted imaging (DWI) sequence. In this phase, which measured the ability of water molecules to experience unrestricted Brownian motion, an ill-defined 2.3x1.9 cm region with significant restriction was noted in the left hepatic lobe segment II which was concerning for ICC (Figure 2). A CT scan of the chest was done to rule out any metastasis. In light of the suspected diagnosis of cholangiocarcinoma and the patient's preserved liver function, a decision was made to proceed with a surgical resection of the cholangiocarcinoma. An exploratory laparoscopy revealed no signs of metastatic disease, and intraoperative ultrasound was performed. Identification of the lesion in the left lateral lobe was quite difficult due to the surrounding fibrotic changes within the liver parenchyma. A left lateral hepatectomy was planned, and the liver capsule was scored. Due to the inability to manipulate the liver during parenchymal resection, the operation was converted to an open surgery through a midline incision, and a successful left lateral hepatectomy was completed. Final pathology showed a poorly differentiated cholangiocarcinoma forming a mass of 2.8 cm with negative resection margins. No perineural or vascular invasion was noted. Background features of the pathology showed CHF (Figure 3). The patient remained in the hospital for three days and was discharged with no significant complications. The patient was doing well at 12 months follow-up without any deterioration of liver function. CA 19-9 levels have returned to baseline levels. | ||||||
|
| ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
Although cholangiocarcinoma is known to develop in fibropolycystic diseases, development of ICC is rare in CHF. In the previous report, the age range for the four patients found at autopsy to have ICC in the setting of CHF was 33–62, half of the patients were female [9]. Two of these patients also had portal hypertension like the patient reported here, which is thought to be found in up to 70% of patients with CHF [9]. Our patient was found to have cholangiocarcinoma during routine surveillance of his liver for the underlying diagnosis of CHF. Elevated tumor markers raised suspicion of cholangiocarcinoma, and imaging studies helped confirm the diagnostic likelihood. Much exists in literature about the imaging signs of cholangiocarcinoma, but making the diagnosis in the setting of CHF is difficult [6] [10]. However, with a high index of suspicion, it is prudent to pursue diagnostic cross-sectional imaging, preferably MRI with DWI. The overall prognosis of patients who develop ICC is poor with an average five-year survival rate of 5–10% [8]. Surgical resection provides the only option for a curative approach. In this patient, the surgical margins were widely negative and no adjuvant therapy was given due to lack of evidence showing demonstrating significant patient benefit from additional treatment. The pathophysiology behind this rare presentation is currently unknown. This could be due to genetic abnormalities of epithelial cells of CHF coupled with other factors such as disturbances of bile flow. Bacterial infection can also serve as a trigger point. Future research to find other patients with overlapping CHF and ICC and to determine the pathophysiology of this condition is crucial to advancing understanding and treatment of patients who present with both conditions. CHF and its associated syndromes have been linked with a variety of different genes. In murine models, it was found that angiotensin converting enzyme (ACE) levels and TGF-β expression were increased in mice with CHF [11]. CHF is the main feature of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma and hepatic fibrosis, also known as COACH syndrome. Studies on COACH syndrome have shown gene involvement in MKS3/TMEM67 genes [12] [13]. Exome sequencing on patients with Caroli syndrome have shown association with WDR19 mutations [14] [15]. While these genes currently do not play a significant role in diagnosing or treating this condition, there may be clinical utility in the future of these genetic studies. Additionally, knowledge of individual gene targets may one day lead to possible therapies. CHF is an inherited malformation with haphazardly proliferating bile ducts and a variable degree of periportal fibrosis; it is characterized by portal hypertension, hepatic fibrosis, and renal cystic disease [2] [6] [7]. The clinical picture of CHF can vary; however, the usual presentation is in childhood or early adulthood with symptoms involving other organ systems. The majority of patients are asymptomatic with the major finding on physical exam being hepatomegaly. This patient was not transplanted due to ICC not being an indication for orthotopic liver transplant. However, Klatskin tumor, an extra-hepatic cholangiocarcinoma, is an indication for liver transplant. Liver transplantation appears to be an effective treatment for perihilar cholangiocarcinoma but there is currently no widespread use of orthotopic liver transplantation for ICC [16]. | ||||||
|
Conclusion
| ||||||
|
All patients with congenital hepatic fibrosis should be monitored for the development of cholangiocarcinoma. If the patient has compensated disease, treatment via partial hepatectomy is preferred. With vigilant screening and appropriate treatment, it is possible to decrease morbidity and mortality from the development of congenital hepatic fibrosis in the setting of congenital hepatic fibrosis. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Adeel S. Zubair – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Ali Metwaly – Analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published Justin M. Burns – Analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published John A. Stauffer – Analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2015 Adeel S. Zubair et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|