| Table of Contents |  |
|
Case Series
|
| CT-guided fine-needle aspiration cytology of solid pseudopapillary tumor of the pancreas: Case series and review of literature |
| Anadi Roy Choudhury1, Bhaskar Mitra2, Palash Bhattacharya1, Aditi Bhattacharya1 |
|
1Assistant Professor, Department of Pathology, RG Kar Medical College & Hospital, Kolkata, West-Bengal, India.
2Assistant Professor, Department of Pathology, Midnapore Medical College & Hospital, Paschim Medinipur, West-Bengal, India. |
|
Article ID: 100007IJHPDARC2012
doi:10.5348/ijhpd-2012-7-CS-5 |
|
Address correspondence to: Dr. Bhaskar Mitra 54/2/G, Feeder Road, P.O- Belgharia, Pin Code - 700056 Kolkata West-Bengal India Phone: +91 9874174040 Email: bhaskarmitra12@gmail.com |
|
[HTML Abstract]
[PDF Full Text]
|
| How to cite this article: |
| Choudhury AR, Mitra B, Bhattacharya P, Bhattacharya A. CT-guided fine-needle aspiration cytology of solid pseudopapillary tumor of the pancreas: Case series and review of literature. International Journal of Hepatobiliary and Pancreatic Diseases 2012;2:20–23. |
|
Abstract
|
|
Introduction:
Solid pseudopapillary tumor is a rare entity considered to be a tumor of low malignant potential and therefore proper evaluation and diagnosis is important.
Case Series: We report two cases of solid pseudopapillary tumor with special emphasis on cytomorphology, histopathology and immunohistochemistry. Preoperative image guided cytopathology is helpful for correct identification of this unusual neoplasm as complete resection is the treatment of choice and is associated with long term survival even in the presence of metastatic disease. Conclusion: Solid pseudopapillary tumor is a distinctive pancreatic neoplasm whose cell phenotype remains a mystery, so we report this case series with extensive literature review. | |
|
Key Words:
Fine-needle aspiration cytology, Solid pseudopapillary tumor, Pancreas
| |
|
Introduction
| ||||||
|
Solid pseudopapillary tumor of the pancreas (SPTP) is a rare neoplasm with a reported frequency of 0.17–2.7% of all nonendocrine tumors of the pancreas, [1] and 6.5% of all pancreatic tumors and tumor-like lesions resected as reported in one large institutional study. [2] It was first described by Frantz et al. and it was also called papillary and solid epithelial neoplasm, papillary-cystic neoplasm and cystic-solid papillary carcinoma until the WHO pancreatic tumor working group termed it as Solid pseudopapillary neoplasm. [3] Though rare it can be diagnosed accurately by Computed Tomography (CT)-guided fine needle aspiration cytology preoperatively as there are some other pancreatic tumors that share the same CT findings of encapsulated and cystic tumor like SPTP. Cytologically the features of SPTP are distinct from these tumors. It is important that this tumor be accurately diagnosed because management protocols differ from other tumor types originating in the pancreas. | ||||||
|
Case Series
| ||||||
|
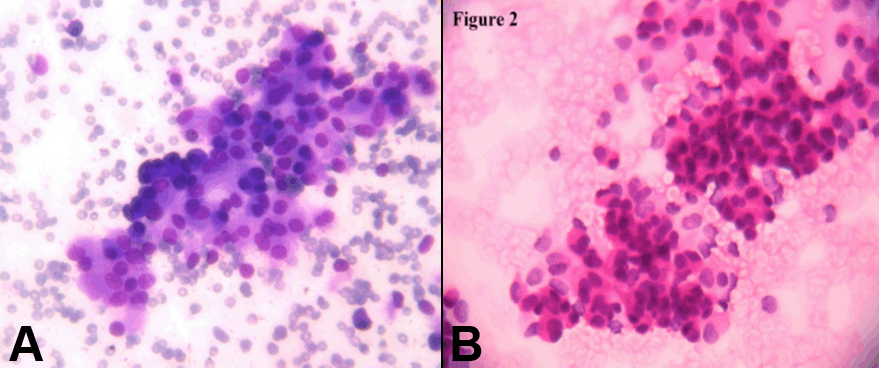
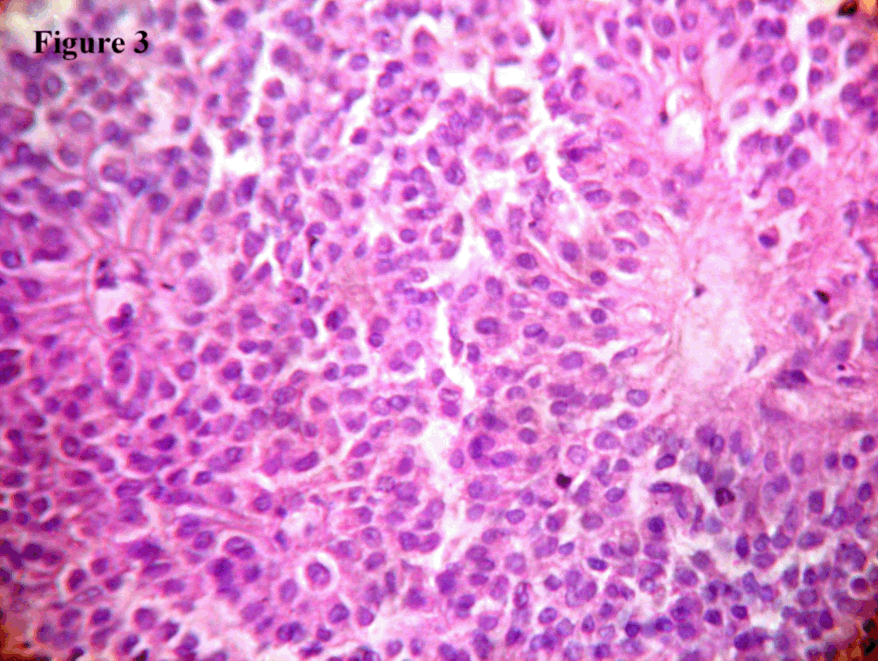
Case 1 Case 2 Cytology smears showed moderate cellularity with cells arranged in loosely cohesive clusters and in papillary fashion. Individual cells were uniform and bland with round to oval nuclei having evenly dispersed or somewhat granular chromatin and small indistinct nucleoli. Mitotic figures and nuclear atypia were absent. The cytoplasm ranged from scant to moderate (Figure 2 A, B). Grossly, both tumours were well circumscribed globular mass with cystic variegated areas on cut section. Adjacent spleen in the first case and duodenum in the second case were not involved. Histologically, the tumor cells were arranged in solid sheets, cords and pseudopapillae. Individual cells were uniform, medium sized, round with eosinophilic cytoplasm and bland nuclei with finely dispersed chromatin and inconspicuous nucleoli and some nuclei showed indentation. Mitotic figures were scanty. Vascular invasion was not noted. Surrounding capsule showed calcification (Figure 3). CD10 immunostain was done which showed focal positivity of moderate intensity. With these classic features a diagnosis of SPTP was offered. | ||||||
| ||||||
|
| ||||||
| ||||||
|
| ||||||
|
| ||||||
|
Discussion
| ||||||
|
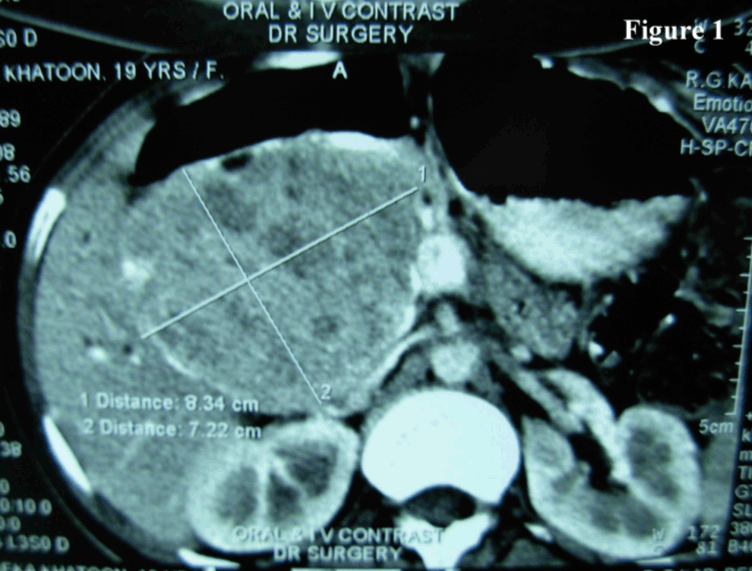
SPTP is a rare tumour occurring predominantly in adolescent girls and young women (mean age 35 years; range 8–67 years). It is rare in men (mean age 35 years; range 25–72 years) without any apparent ethnic preference. [3] Clinically, SPTP may present as an abdominal mass with discomfort or pain or it may be an incidental finding in work up for unrelated conditions. Jaundice is rare even if the tumor arises from the head of the pancreas. [3] These tumors are generally large with a mean diameter of 10.3 cm and approximately 72% arise in the body and tail of the pancreas and less frequently in the head. SPTP may present at extrapancreatic sites such as mesocolon, retroperitoneum, omentum, liver and duodenum. [4] In our case, the sex (female) and age (20 years and 19 years respectively) and tumor localization (tail in the first case and body in the second case) were typical of SPTP. SPTP usually runs a benign course though about 15% cases it may present with local extension into blood vessels and organs, local recurrence and distant metastasis. [5] Most of the tumors are benign appearing but tumors which have the potential to metastasize show cellular atypia. Pathologic features related to metastatic potential include diffuse growth pattern, venous invasion, nuclear pleomorphism, mitotic rate, necrosis and areas of dedifferentiation. [6] Ultrasonography (USG) and computed tomography (CT) scan reveal a sharply demarcated, variably solid and cystic mass without any internal septation. The tumor margin may contain calcifications. Cytogically, it presents with highly cellular aspirates with single cells, loose clusters, and branching papillary fronds. Tumor cell are monotonous and bland with or without clefted nuclei (or nuclear grooves). Myxoid and metachromatic stroma and background material may be found. Necrotic debris is seen rarely when areas of cystic degeneration are sampled. [7] In our cases we did not get the necrotic material in the background. Immunohistochemistry of SPTP has produced conflicting results with regard to tumor cell phenotyping. Patil et al. suggested that SPTP possibly originate from the primordial cells and lack definite exocrine and endocrine differentiation. [8] Notohara et al. and others performed immunohistochemical analysis of 19 cases of SPTPs and showed that all of them exhibited immunoreactivity for CD56 and CD10. Fifteen of them expressed other neuroendocrine markers focally with the exception of Chromogranin A. [9] Another study of three SPTP cases demonstrated immunoreactivity for vimentin, CD10, CD56, synaptophysin and nuclear accumulation of beta catenin. [10] But due to financial constraints we have only done CD10 to confirm our diagnosis and found it to be positive. | ||||||
|
Conclusion
| ||||||
|
A high index of suspicion is necessary to diagnose SPTP. In such cases CT-guided FNAC appears to be of value in preoperative diagnosis. Surgical excision offers the best chance for cure and patients have an excellent prognosis. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions:
Anadi Roy Choudhury - Substantial contributions to conception and design, Acquisition of data, Drafting the article, revising it critically for important intellectual content, Final approval of the version to be published Bhaskar Mitra - Substantial contributions to conception and design, Analysis and interpretation of data, Interpretation of Immunohistochemistry data, Drafting the article, Critical revision of article, Final approval of the version to be published Palash Bhattacharya - Substantial contributions to conception and design, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Aditi Bhattacharya - Substantial contributions to conception and design, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published. |
|
Guarantor of submission:
The corresponding author is the guarantor of submission. |
|
Source of support:
None |
|
Conflict of interest:
Authors declare no conflict of interest. |
|
Copyright:
© Anadi Roy Choudhury et al. 2012; This article is distributed the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any means provided the original authors and original publisher are properly credited. (Please see Copyright Policy for more information.) |
|
|